주문제작 서비스 문의
찾으시는 제품이 저희 사이트에 없으신가요? 단백질 주문 제작 서비스를 이용해보세요!
주문제작 서비스 문의 >>






















 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!

Cell and Gene Therapy (CGT) made its entrance as a potential treatment for blood-based cancers through the FDA’s accelerated approval process. Despite its introduction as the next-generation of biologic-based treatments, these pioneering medicines have been quickly reined in by the dynamic landscape of regulatory guidelines that are designed to ensure patient safety.
Guiding principles sent from regulatory agencies in the United States and Europe have been published to regulate various facets of this rapidly growing field. Within this intricate regulatory framework, securing Investigational New Drug (IND) approval for CGT products can be a challenge.
Something that is overlooked when it comes to obtaining IND approval is ensuring that the raw materials utilized are manufactured under the necessary quality controls behind the raw materials used. When it comes to our Good Manufacturing Practice (GMP) products, we performed extensive research on the global regulations and standards pertaining to raw or ancillary materials before production. This means from product inception to commercialization, consideration of regulatory requirements have been incorporated into the material design. Therefore, our ancillary materials meet the highest GMP standards mandated by regulatory bodies across the globe.
So which regulatory requirements did we consider for our GMP-grade products?
Across industry, GMP-grade products are expected to adhere to the following pharmacopeia documents, governing different aspects of raw materials.
1. USP<1043>Ancillary Materials for Cell, Gene and Tissue-Engineered Products
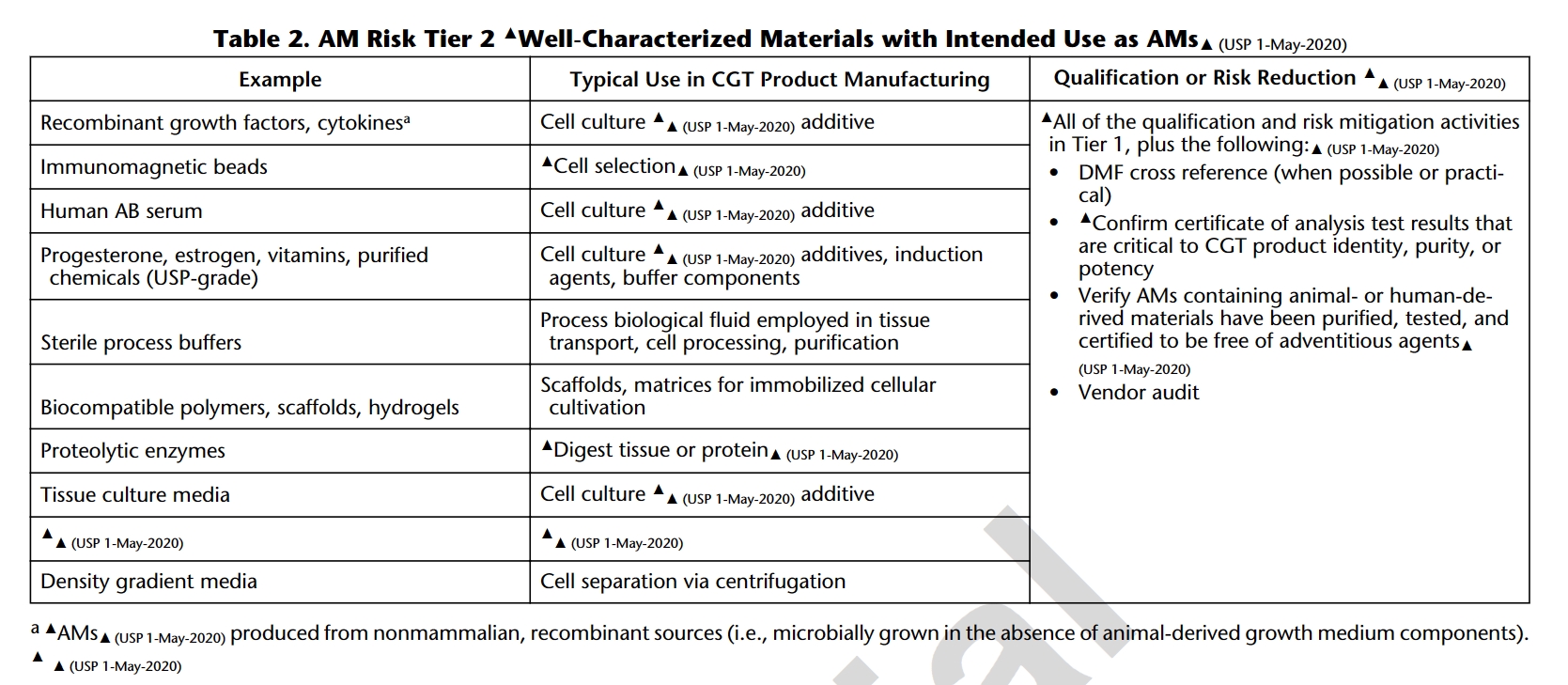
USP 1043 classifies ancillary materials (AM) into four tiers based on their significance in Cell and Gene Therapy (CGT) processes and risk assessment. Essential documentation, including Drug Master Files (DMF), Certificates of Analysis (CoA), and assurances that AM with animal or human-derived materials has been purified, tested, and is free of external exogenous factors are highlighted. As such, most cytokines for cell culture additives fall into Tier 2 where regulations play a crucial role in guaranteeing the traceability, quality, and safety of ancillary materials, emphasizing transparency and reliability in CGT manufacturing processes.
2. IPRP (International Pharmaceutical Regulators Programme) “General Considerations for Raw Materials Used in the Production of Human Cell and Gene Therapy Products”
• Establishing a Quality Management System
The IPRP report, in Section 3.1, mandates CGT manufacturers to institute a robust quality management system, encompassing raw material certification programs. This encompasses essential facts such as supplier approval, execution of quality agreements, provision of CoA, and submission of supporting documents. As such, responsibility falls upon the CGT manufacturer to perform due diligence when it comes to finding raw materials supplier. This means inspecting the adherence of raw material storage requirements, stability control, and the effective management of expiration dates by both supplier and manufacturer.
• Ensuring Transparency of Origin and Traceability
Section 3.3 underscores the significance of origin and traceability considerations. Raw materials must be accompanied by pertinent documents, including Certificates of Origin (COO), facilitating traceability and identification of the origin of materials or components used in the production process. When utilizing materials from human or animal sources, CGT product manufacturers are advised to selectively opt for materials with accessible source information. This may necessitate establishing agreements with suppliers to provide materials sourced from specific regions, ensuring a transparent and traceable supply chain.
• Microbial Safety in Raw Material Production
In addressing microbial safety concerns during raw material production (Section 4.2), the report highlights a key issue: most CGT products do not undergo terminal sterilization. This means rigorous sterile testing of raw materials used in CGT product production is imperative. Given the limitations of traditional virus inactivation/removal procedures for CGT products, the report advocates for the use of validated methods for virus inactivation/removal steps when utilizing biologically sourced raw materials. In cases where such procedures are impractical, comprehensive testing for the presence of relevant viruses in raw materials should be conducted, accompanied by a well-founded rationale for their utilization. This multifaceted approach ensures the robustness and safety of raw materials integral to the production of innovative Cell and Gene Therapy products.
3. European Pharmacopeia 5.2.12 “Raw materials for the production of cell-based and gene therapy medicinal products”
This pharmacopeia document outlines the stringent requirements for raw materials of biological origin employed in the production of CGT products across their lifecycle within the EU region. Specifically, in Section 3.2 addressing production, key considerations include the imperative for production within an appropriate quality management system and facility, incorporating a stringent aseptic production process that considers the impact of additives, notably antibiotics.
General quality requirements for raw materials encompass a comprehensive set of criteria, covering identification, purity, biological activity, impurities (including residual host cell protein/DNA), microorganisms, virus contaminants, mycoplasmas, water purity, content, and biological activity (specific activity). These criteria collectively ensure the integrity and safety of raw materials throughout the production process.
A pivotal emphasis is placed on the sterility of raw materials, with a mandate that they are either produced as sterile or terminally sterilized under aseptic conditions unless explicitly specified otherwise. In cases where the raw material is not inherently sterile, a prerequisite is the knowledge and disclosure of the level of microbial contamination. This detailed approach to raw material specifications aligns seamlessly with the overarching goal of ensuring the highest standards in quality and safety throughout the CGT manufacturing process.
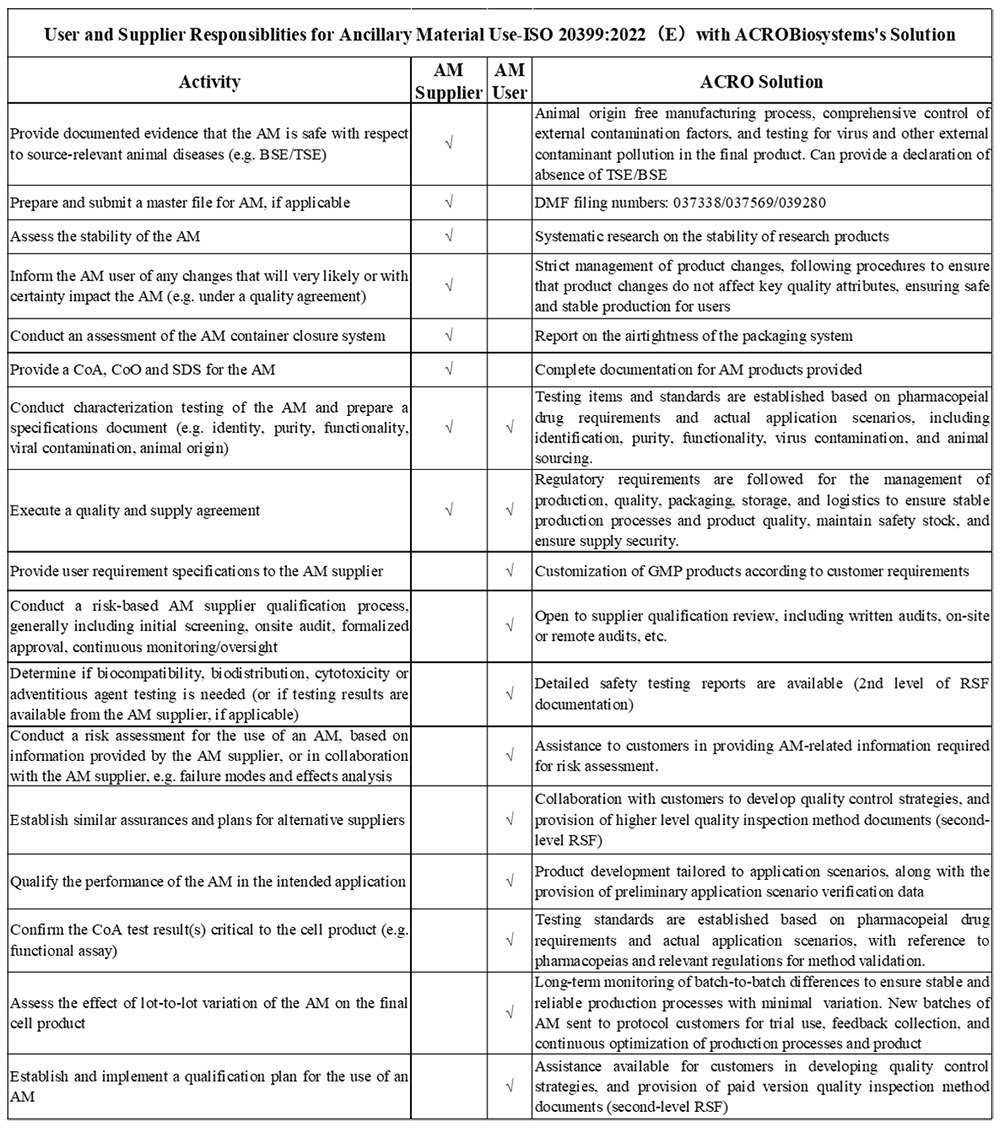
4. ISO 20399:2022 (E) “Biotechnology — Ancillary materials present during the production of cellular therapeutic products and gene therapy products”
The International Organization for Standardization introduced ISO 20399:2022(E), offering a comprehensive framework that describes the roles of AM suppliers and users in the production of CGT products.

Due to the complex interchange between supplier and manufacturer, we are dedicated to streamlining this process. We offer tailored solutions to assist and support our valued customers in navigating these requirements seamlessly. Our commitment to staying on top of industry regulations ensures that we can provide the necessary support to facilitate a smooth and compliant production process.
Having thoroughly examined key guidelines established by regulatory bodies, we are able to effectively support our customers regulatory filing requirements in both the United States and the EU. Here are a few ways in which we can be a valuable partner in navigating the regulatory landscape for CGT production.
a. Comprehensive Regulatory Support Documents:
At the forefront of our capabilities is the careful compilation of Regulatory Support Files (RSF), aligning seamlessly with international regulations and specific customer or official requirements. These documents meticulously outline the indispensable qualifications required for GMP-grade materials in CGT production, custom-tailored to meet the diverse needs of customers across various developmental stages.
Level 1 Documents:
The Level One documents, focusing on product qualifications, are provided free of charge to customers who have purchased GMP products. These documents, approximately 40-60 pages, are designed to serve the needs of customers in the research and pre-clinical stages. They act as essential proof of supplier qualifications during the material screening phase.

Level 2 Documents:
Level Two documents are defined as unique quality and safety documents, comprising over 1000 pages. These documents are available for purchase and target customers entering the clinical phase. At this stage, customers require detailed information on raw materials to support their clinical applications or market authorization. Level Two documents include thorough Standard Operating Procedures (SOPs), analytical method validation reports, and contribute significantly to saving time, costs, and manpower in material analysis development and validation.

b. Comprehensive GMP Products with U.S. FDA Filing Support:
We actively support Drug Master File (DMF) filing, a comprehensive set of documents containing information on product chemistry, manufacturing, and controls (CMC) information. As the first company to secure FDA DMF filing for recombinant protein reagents, we continue that support across our GMP products and other recombinant proteins.
c. IND/New Drug Application (NDA)/Biological License Application (BLA) Stage Compliance Support:
We also provide on-site support to customers throughout the IND/NDA/BLA filing process. This support encompasses addressing regulatory queries related to ancillary materials, ensuring that our customers have the correct documentation needed for their filings.
The article delves deep into the intricate regulatory landscape of Cell and Gene Therapy (CGT) manufacturing, with a primary focus on ensuring compliance with the stringent regulatory requirements of both the US and EU. Understanding the supplier-manufacturing relationship and the necessary checks and balances behind manufacturing cell therapies can greatly streamline IND approval. This examination of key regulatory guidelines, encompassing USP, IPRP, European Pharmacopeia, and ISO, sheds light on the criteria governing ancillary materials while highlighting steps that we take help navigate the intricate regulatory landscapes of CGT production, while catering to the diverse needs of customers across developmental stages.





This web search service is supported by Google Inc.
















 A-Z
A-Z





















