주문제작 서비스 문의
찾으시는 제품이 저희 사이트에 없으신가요? 단백질 주문 제작 서비스를 이용해보세요!
주문제작 서비스 문의 >>






















 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!

細胞療法製品の製造には無菌的な性質があるため、製造プロセスで使用する原材料も無菌状態であることが不可欠です。無菌性を保証できないことは、細胞療法製品の製造における危機的な失敗を意味し、品質要件に適合する医薬品を適切な時期に患者に届けられない原因となります。
そのため、原材料の無菌性は、細胞療法の観点において極めて重要な品質特性として際立っています。原材料の製造プロセスで採用されている無菌性管理戦略は、特にサイトカイン、抗体、酵素の製造プロセスにおいて重要性が高いと考えられます。これらのプロセスには、多くの場合、最終非滅菌プロセスが必要となります。徹底したリスクアセスメントを実施して潜在的な微生物汚染源を特定することが重要です。リスクアセスメントには、設備、装置、プロセス設計、マテリアルハンドリング、人員に関する要求事項、製造作業、環境モニタリングなどのさまざまな要因が含まれますがこれらに限定されず、その要因の包括的な検討を含む必要があります。続いて、製造管理と品質管理を共に強化するための、対象を限定した管理措置を策定することが不可欠です。
上記を踏まえ、無菌性管理戦略は、各国のさまざまな医薬品の製造管理、品質管理に関する基準 (GMP) の規制およびガイドラインによって定められた厳格な基準に従います。医薬品業界は、以下を含め、一般的に認知されている一連の GMP 規制およびガイドラインを一貫して遵守します。
1. 欧州連合における医薬品および医薬部外品の製造管理および品質管理に関する基準 (EU GMP) 付属書 1: Manufacture of Sterile Medicinal Products (無菌医薬品の製造)
2. 米国 FDA 現行の医薬品の製造管理、品質管理に関する基準 (21 CFR Part 210、211、600)
3. FDA 業界向けガイダンス: Sterile Drug Products Produced by Aseptic Processing (無菌操作法で製造される無菌製剤)
4. PIC/S GMP 付属書 1: Manufacture of Sterile Medicinal Products (無菌医薬品の製造)
Aこれらの中でも、2023 年 8 月 25 日に採択された EU GMP 付属書 1 の新版には、無菌製品の製造に関する特に厳格な要件が記載されています。この規制は、特に、原薬、医薬品添加剤、一次包装材料、最終剤形、さらには多様な包装容量、製造プロセス、技術を対象としています。その一方で、この規制には、品質リスクマネジメント (QRM) の原則に従って、すべての無菌製品の製造に使用する設備、装置、システム、および手順の全般的な設計および管理に関する一般指針が含まれています。その目的は、最終製品に微生物、粒子状物質、エンドトキシンまたは発熱性物質による汚染がないことを保証することです。このガイダンスでは、設備、装置、プロセス、材料、試験、および環境モニタリングなどの側面からの、全体的な評価と汚染管理を重視しています。
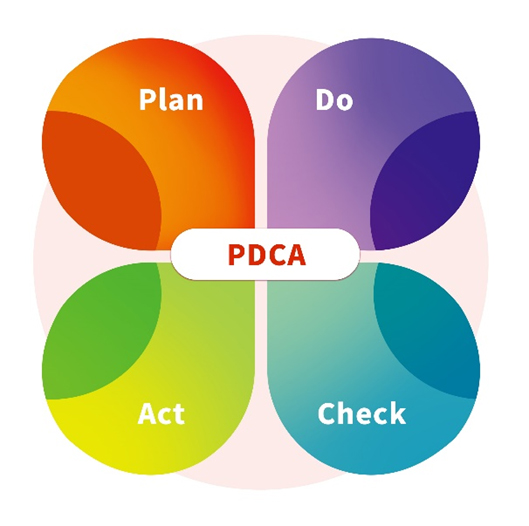
ACROBiosystems は、GMP Gradeの細胞療法製品が前述の規制およびガイダンス基準に適合することを保証しています。当社は、総合的な手法を採用し、設備、装置、材料、プロセス、および人員に関する無菌性管理戦略を綿密に設計し、取り入れています。継続的に改善し強化することが、ACROBiosystems の品質保証方針に不可欠であり、PDCA (Plan (計画)、Do (実行)、Check (検証)、Act (改善)) ツールを体系的に適用することで円滑に進めています。
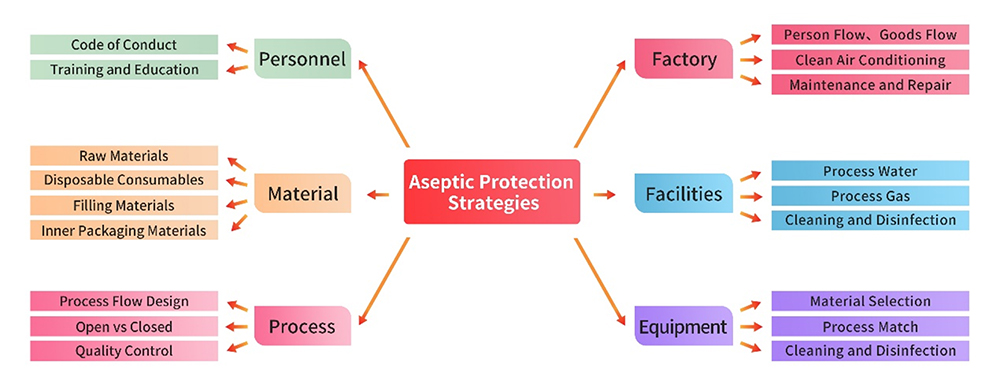
1. 設備および装置
製造設備、ユーティリティ、装置は、厳格な GMP 規制に準拠しています。製品の製造を開始する前に、各設備と装置に対し包括的なギャップ分析およびリスクアセスメントが実施されます。用法、洗浄、消毒、保全などの領域を網羅する手順書を作成し、厳格に実施しています。上記の措置により、これらのインフラストラクチャの管理を正確に実行し、無菌性管理に対する悪影響を防止しています。
2. 人員管理
製造および品質マネジメント活動に従事する人員には、製造規格と汚染および交差汚染の効果的管理を共に維持するために必要な学歴、訓練、および経験が必要です。このような人員の適格性に対する取り組みにより、製造プロセスの完全性を維持するために重要な作業を、円滑かつ確実に実行できます。
3. 原材料の管理
製造に使用する原材料および補助材料は、包装材料を含め、医薬品Gradeの材料を調達することが望まれます。厳格なサプライヤ マネジメント業務を、適格性確認の収集、現地監査などのプロセスを通じて実施します。品質管理戦略を、特に微生物管理の領域において包括的なリスクアセスメントを基に綿密に策定し、製造プロセスで使用する原材料の完全性を確保します。
4. 製造プロセスと品質管理
製造プロセスにおける無菌性管理の重要なポイントは以下のとおりです。
| 製造環境 | 原材料の調製: ISO クラス C および A 製剤の製造: ISO クラス B および A 動的環境モニタリング |
| 溶液およびプロセスガス | 0.1 μm/0.22 μm 滅菌ろ過 二次滅菌ろ過による製剤の製造 フィルター使用の前後での完全性試験 |
| シングルユース消耗品 | ガンマ線滅菌 使用前の完全性確認 |
| プロセス管理 | 無菌的接続 エンドトキシン管理 無菌プロセス シミュレーション充填 |
| 装置管理 | 再利用可能な装置の洗浄プロセスと洗浄確認 |
| 洗浄および消毒 | 消毒効果のベリフィケーション |
| 品質管理 | 原材料の微生物限度管理 製品の無菌試験は USP に準拠しています |
上流の細胞培養:
• 細胞培養の生産段階では、厳格な規制を人員の服装および行動要件に適用して、作業による汚染リスクを最小限に抑えます。
• 細胞の回収とフラスコによる拡張培養段階は、周辺環境としてクラス C の清浄区域で行われます。開放系の操作は、動的環境モニタリングを同時に実施できる層流保護下のクラス A 安全キャビネット内で行われます。確実に無菌状態にするため、リアクターの拡張培養および灌流培養ステージにはシングルユース閉鎖系システムを取り入れ、接種、栄養供給、その他のプロセスに無菌溶接技術を採用して、微生物汚染を効果的に防ぎます。
• 細胞培養段階では、使用する原材料を厳格にエンドトキシン管理し、厳しい品質出荷判定基準に合格した後にのみ、これらの原材料を使用できます。培地および溶液の調製は、注射用水に関する薬局方の要件に準拠しています。調製後、溶液を、0.1 μm/0.22 μm のフィルターに通して滅菌し、フィルターの完全性を検査します。同時に、無菌試験を実施して、両方の試験で要件を満たす結果が得られた場合にのみ、溶液を細胞培養生産に使用できます。このプロセスで使用する二酸化炭素、酸素、圧縮空気などのガスは、高純度または食添Gradeの基準を保っています。リアクターに接続するプロセスガスを、0.22 μm フィルターに通して効果的に滅菌します。反応バッグや保存バッグなどのシングルユース消耗品は、無菌性を確保するために放射線滅菌され、液体と直接接触する材料に微生物汚染がないことを保証します。
• 清澄化ろ過に使用する深層ろ過膜はシングルユースです。ろ過膜の包材を、使用前に注射用水で洗浄し、包材に含まれる不純物を除去します。清澄化ろ過の後、下流の精製プロセスに入る前に、0.22 μm フィルターによる 2 回目の液体のろ過を実施して、微生物的負荷を効果的に低減します。下流の精製に移る前に、中間製品がプロセス要件を満たすことを確認するため、回収した清澄化液体のエンドトキシン試験を実施します。
下流の精製:
• 精製プロセスで使用する分離カラムとクロマトグラフィー担体は個々のプロジェクト専用とし、異なる製造段階にまたがる交差汚染リスクを防止します。各バッチの製造後、クロマトグラフィー システムを徹底的に洗浄し、リスクアセスメントに基づき清浄度を確定します。クロマトグラフィー担体を、手順書に従って洗浄し、適切に保管し、次のバッチ製造に使用する前に洗浄して、微生物による影響を効果的に低減します。精製段階における、製品と直接接触する材料および消耗品の微生物管理方法は、細胞培養段階の方法と同様であり、微生物の混入を確実に防止できます。再利用可能な装置や機器、器具は、無菌性を保つために湿熱滅菌されます。
• 分離およびクロマトグラフィー後の液体は、ナノろ過および限外ろ過プロセスを経て、最終的に 0.2 μm のフィルターによる滅菌ろ過に至ります。ろ過後、環境に由来する微生物汚染を軽減するため、Grade A の層流保護下で液体を充填します。このバルク液体に、品質基準に従って、微生物限度試験やエンドトキシン評価などのさまざまな試験を実施します。これらの試験に合格し、品質部門の承認を受けた後、液体を最終製品の充填に進めます。
製剤:
• 最終製品の充填は、最終非滅菌の無菌的製造プロセスに従って進み、厳格な B+A 製造環境を確保します。滅菌ろ過後、半製品が得られます。アセンブリ、充填、施栓、自動装入取出し、凍結乾燥は、Grade A の無菌環境 (PMS 連続環境モニタリング システム) 内で完全動化装置とシングルユース無菌充填システムを使用して実施されます。凍結乾燥後、C+A 環境で密封作業を完了します。包装製品の手作業による目視検査、ラベル貼付、および追加の作業を実施します。これらは最終製品専用の保管場所に置かれ、検査後に出荷されます。
• 厳格な無菌プロセスのバリデーションには、APS (培地シミュレーションバイアル) 内での無菌栄養培地や製品代用品の使用を含む、無菌プロセス管理による定期的な確認が取り入れられています。APS では、材料の滅菌および洗浄から容器の密封までに行われるすべての無菌操作を評価します。信頼性の高い培養結果を保証するため、培地に対し培地性能試験を実施します。微生物侵入試験は、製品包装システムの密閉性を確保する手段として用いられ、保管中の製品の無菌性を保証します。
• 最終製品の品質管理: 滅菌試験は、一連の重要な管理措置の中で、無菌性を確保するための最終ステップです。無菌試験を無菌アイソレータ内で実施します。無菌試験のサンプリングには、表示を目的とするバッチ充填の開始時と終了時の製品が含まれます。無菌性の分析方法は薬局方の要件に厳密に準拠しており、ATCC および CMCC 指定菌株を使用し、生物学的製剤の培養に関する薬局方の要件に従って実施し、結果の信頼性を保証します。
要約すると、ACROBiosystems の GMP Grade製品は、包括的な無菌的保護戦略、厳格な品質管理、および GMP 規則に遵守して製造されています。これらの無菌プロセスは、CGT 製品の登録および商品化に対応します。
• 細胞・遺伝子治療に重要な原材料の製造における外部汚染物質の制御。
• CGT の不可欠な原材料を製造するための無菌生産体制
• CGT における重要な原材料の品質管理システム
• CGT における重要な原材料のグローバルサプライチェーンセキュリティシステム
• CGT の重要な原材料が米国規制要件を適切に満たす方法
• ...

This web search service is supported by Google Inc.
















 A-Z
A-Z























