주문제작 서비스 문의
찾으시는 제품이 저희 사이트에 없으신가요? 단백질 주문 제작 서비스를 이용해보세요!
주문제작 서비스 문의 >>






















 Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.  Limited Edition Golden Llama is here! Check out how you can get one.
Limited Edition Golden Llama is here! Check out how you can get one.
Offering SPR-BLI Services - Proteins provided for free! Offering SPR-BLI Services - Proteins provided for free!
Here come GMP Grade Cytokines!Free Sample is available! Here come GMP Grade Cytokines!Free Sample is available!
> Insights > Sonderthema zur tiefgreifenden Interpretation der Qualität von GMP-Produkten - Thema 2 
Angesichts des aseptischen Charakters der Herstellung von Zelltherapieprodukten ist es zwingend erforderlich, dass die für den Prozess verwendeten Rohstoffe steril sind. Das Versagen bei der Sicherstellung der Sterilität stellt einen entscheidenden Rückschlag bei der Herstellung von Zelltherapieprodukten dar und führt dazu, dass Medikamente, die den Qualitätsanforderungen entsprechen, nicht rechtzeitig an Patienten abgegeben werden können.
Daher stellt die Sterilität ein ausschlaggebendes Qualitätsmerkmal für Rohstoffe im Zusammenhang mit der Zelltherapie dar. Die aseptische Kontrollstrategie bei der Herstellung von Rohstoffen gewinnt vor allem bei der Produktion von Zytokinen, Antikörpern und Enzymen an Bedeutung. Bei diesen Verfahren werden häufig nicht-terminale Sterilisationsverfahren eingesetzt. Die Durchführung einer gründlichen Risikobewertung ist von entscheidender Bedeutung, um potenzielle Quellen mikrobieller Kontamination zu ermitteln. Die Bewertung sollte eine umfassende Prüfung verschiedener Faktoren beinhalten, einschließlich, aber nicht beschränkt auf die Anlage, die Ausrüstung, die Prozessgestaltung, die Materialhandhabung, den Personalbedarf, die Produktionsabläufe und die Umweltüberwachung. In der Folge ist es wichtig, gezielte Kontrollmaßnahmen zu erstellen, um sowohl das Produktionsmanagement als auch die Qualitätskontrolle zu verbessern.
In diesem Zusammenhang unterliegen aseptische Kontrollstrategien strengen Standards, die durch verschiedene GMP-Vorschriften und -Richtlinien in verschiedenen Ländern festgelegt sind. Die Industrie hält sich konsequent an eine Reihe von allgemein anerkannten GMP-Vorschriften und -Richtlinien, die Folgendes umfassen:
1. Gute Herstellungspraxis der Europäischen Union (EU-GMP) Anhang 1: Herstellung von sterilen Arzneimitteln
2. Aktuelle Gute Herstellungspraxis der amerikanischen FDA (21 CFR Teil 210, 211, 600)
3. FDA-Leitfaden für die Industrie: Sterile Arzneimittelprodukte, die durch aseptische Verarbeitung hergestellt werden
4. PIC/S GMP Anhang 1: Herstellung von sterilen Arzneimitteln
AInnerhalb dieser Leitfäden enthält die neue Fassung von Anhang 1 der EU-GMP, die am 25. August 2023 eingeführt wurde, die strengsten Anforderungen für die Herstellung steriler Produkte. Die Verordnung deckt insbesondere aktive pharmazeutische Wirkstoffe, Hilfsstoffe, Primärpackmittel, fertige Darreichungsformen sowie verschiedene Verpackungsgrößen, Produktionsverfahren und Technologien ab. Gleichzeitig enthält die Verordnung allgemeine Leitlinien für die Gesamtkonzeption und -kontrolle von Einrichtungen, Ausrüstungen, Systemen und Verfahren, die für die Herstellung aller steriler Produkte verwendet werden und folgt dabei den Grundsätzen des Qualitätsrisikomanagements (QRM). Ziel ist es, sicherzustellen, dass das Endprodukt frei von Mikroorganismen, Partikeln und Endotoxin-/Pyrogenkontaminationen ist. Der Leitfaden legt den Schwerpunkt auf die Gesamtbewertung und Kontaminationskontrolle unter Aspekten wie Anlage, Ausrüstung, Verfahren, Material, Prüfung und Umweltüberwachung.
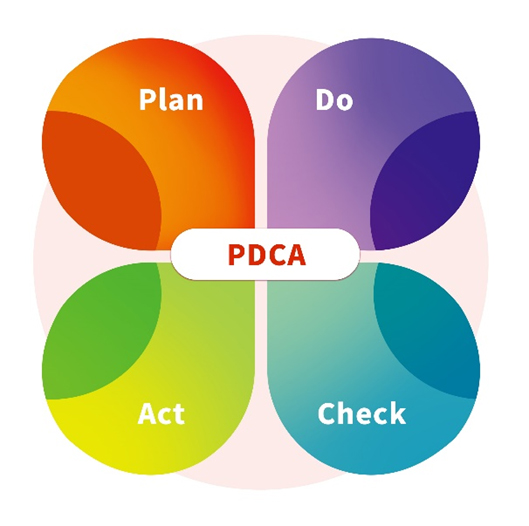
ACROBiosystems stellt sicher, dass seine Zelltherapieprodukte in GMP-Qualität mit den oben genannten regulatorischen Standards und Leitlinien übereinstimmen. Mit einem integrierten Ansatz entwirft und integriert das Unternehmen sorgfältig aseptische Kontrollstrategien für Anlagen, Ausrüstung, Materialien, Prozesse und Personal. Die kontinuierliche Verbesserung und Weiterentwicklung sind integraler Bestandteil des Engagements von ACROBiosystems für die Qualitätssicherung, die durch die systematische Anwendung von PDCA-Instrumenten (Plan-Do-Check-Act) unterstützt wird.
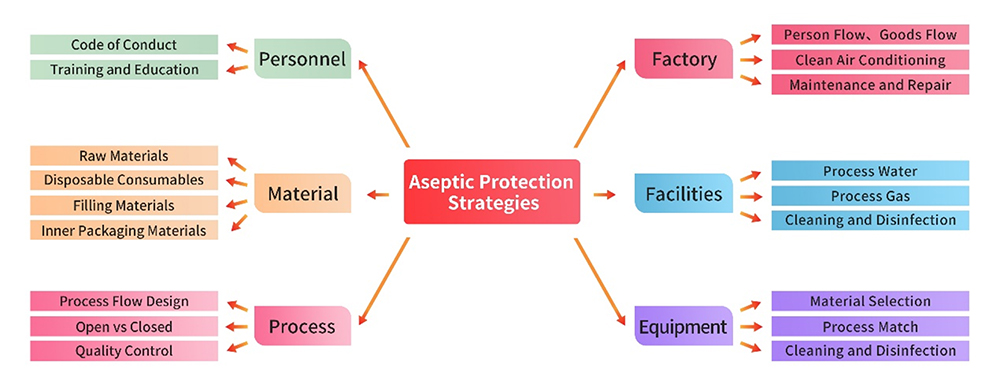
1. Einrichtungen und Ausrüstung
Die Produktionsanlage, die Versorgungseinrichtungen und die Ausrüstung entsprechen den strengen GMP-Vorschriften. Vor der Aufnahme der Produktion werden für jede Anlage und jedes Gerät eine umfassende Abweichungsanalyse und Risikobewertung durchgeführt. Es werden schriftliche Verfahren entwickelt und strikt durchgesetzt, die Bereiche wie Nutzung, Reinigung, Desinfektion und Wartung abdecken. Diese Maßnahmen stellen sicher, dass die Verwaltung dieser Infrastrukturen präzise durchgeführt wird, um nachteilige Auswirkungen auf die aseptische Kontrolle zu vermeiden.
2. Personalmanagement
Das Personal, das in der Produktion und im Qualitätsmanagement tätig ist, verfügt über die erforderliche Ausbildung, Schulung und Erfahrung, um sowohl den Produktionsstandard aufrechtzuerhalten als auch die wirksame Kontrolle von Kontamination und Kreuzkontamination vorzunehmen. Diese Verpflichtung zur Personalqualifikation erlaubt die reibungslose und zuverlässige Ausführung aller Schritte, die für Gewährleistung der Richtigkeit des Produktionsprozesses maßgeblich sind.
3. Materialwirtschaft
Die für die Herstellung verwendeten Roh- und Hilfsstoffe werden vorzugsweise in pharmazeutischer Qualität beschafft, einschließlich der Verpackungsmaterialien. Strenge Praktiken des Lieferantenmanagements werden durch Verfahren wie die Erfassung von Qualifikationen, Audits vor Ort usw. umgesetzt. Qualitätskontrollstrategien werden insbesondere im Bereich der mikrobiologischen Kontrolle auf der Grundlage einer umfassenden Risikobewertung sorgfältig entworfen, um die Zuverlässigkeit der im Produktionsprozess verwendeten Materialien zu gewährleisten.
4. Produktionsprozess und Qualitätskontrolle
Zu den wichtigsten Punkten der aseptischen Kontrolle im Produktionsprozess gehören:
| Production Environment | Raw Material Preparation: ISO Class C and A Drug Product Production: ISO Class B and A Dynamic environment monitoring |
| Solutions and Process Gases | 0.1μm / 0.22μm sterilizing filtration Drug product production with secondary sterilizing filtration Integrity testing before and after filter use |
| Single-Use Consumables | Gamma irradiation sterilization Integrity check before use |
| Process Control | Aseptic connections Bacterial endotoxin control Aseptic process simulation filling |
| Equipment Control | Cleaning processes and cleaning confirmation for reusable equipment |
| Cleaning and Disinfection | Disinfectant efficacy verification |
| Quality Control | Microbial limits control for raw materials Sterility testing of products complies with USP |
Vorgelagerte Zellkultivierung:
• In den Produktionsphasen der Zellkulturen gelten strenge Vorschriften für die Kleidung und das Verhalten des Personals, um das Kontaminationsrisiko durch die Arbeitsabläufe zu minimieren.
• Zellrückgewinnung und Kolbenerweiterungsstufen werden in einer Reinumgebung der Klasse C als Hintergrundumgebung durchgeführt. Offene Arbeitsabläufe werden in einer Biosicherheitskabine der Klasse A unter Laminarschutz durchgeführt, wobei gleichzeitig eine dynamische Umgebungsüberwachung stattfindet. Um aseptische Bedingungen zu gewährleisten, sind die Reaktorexpansions- und Perfusionskulturstufen mit geschlossenen Einwegsystemen ausgestattet, bei denen sterile Schweißtechniken für Prozesse wie Inokulation, Fütterung und andere Vorgänge eingesetzt werden, um eine mikrobielle Kontamination wirksam zu vermeiden.
• Während der Zellkulturphase werden die verwendeten Materialien einer strengen Endotoxin-Kontrolle unterzogen und nur dann verwendet, wenn sie strenge Qualitätsfreigabekriterien erfüllen. Bei der Zubereitung von Nährmedien und Lösungen werden die Anforderungen des Arzneibuchs für Injektionswasser eingehalten. Nach der Zubereitung werden die Lösungen durch 0,1μm/0,22μm-Filter sterilisiert und die Unversehrtheit der Filter wird geprüft. Gleichzeitig werden Sterilitätstests durchgeführt und nur wenn beide Tests zufriedenstellende Ergebnisse liefern, können die Lösungen für die Zellkulturproduktion verwendet werden. Die im Prozess verwendeten Gase, wie Kohlendioxid, Sauerstoff und Druckluft, müssen hochreine oder lebensmitteltaugliche Standards erfüllen. Die an den Reaktor angeschlossenen Prozessgase werden durch einen 0,22μm-Filter wirksam sterilisiert. Einweg-Verbrauchsmaterialien, einschließlich Reaktions- und Lagerungsbeutel, werden durch Bestrahlung sterilisiert, um die Sterilität zu gewährleisten und sicherzustellen, dass Materialien, die in direktem Kontakt mit der Flüssigkeit stehen, nicht durch Mikroorganismen kontaminiert werden.
• Tiefenfiltrationsmembranen für die Klärfiltration sind für den einmaligen Gebrauch bestimmt. Vor der Verwendung werden die Membranverpackungen mit Injektionswasser gespült, um darin enthaltene Verunreinigungen zu entfernen. Nach der Klärfiltration wird die Flüssigkeit einer zweiten Filtration durch einen 0,22μm-Filter unterzogen, um die mikrobielle Belastung wirksam zu reduzieren, bevor sie in die nachgeschalteten Reinigungsprozesse gelangt. Vor dem Übergang zur nachgeschalteten Reinigung wird die gewonnene geklärte Flüssigkeit einem Endotoxintest unterzogen, um sicherzustellen, dass das Zwischenprodukt die Prozessanforderungen erfüllt.
Nachgeschaltete Reinigung:
• Die Trennsäulen und chromatographischen Trägersubstanzen, die im Reinigungsprozess verwendet werden, sind ausschließlich für das jeweilige Projekt bestimmt, so dass das Risiko einer Kreuzkontamination über verschiedene Produktionsstufen hinweg vermieden wird. Nach der Produktion der jeweiligen Charge wird das chromatographische System einer Tiefenreinigung unterzogen, wobei die Reinheit auf der Grundlage einer Risikobewertung bestätigt wird. Chromatografische Trägersubstanzen werden gemäß schriftlicher Vorgaben gereinigt und angemessen gelagert und anschließend vor der Verwendung in der nächsten Produktionscharge gereinigt, um die Auswirkungen von Mikroorganismen wirksam zu verringern Mikrobiologische Kontrollmethoden für Materialien und Verbrauchsmaterialien, die während der Reinigungsphase in direktem Kontakt mit den Produkten stehen, entsprechen denen in der Zellkulturphase, so dass die Einschleppung von Mikroorganismen verhindert wird. Wiederverwendbare Geräte und Instrumente werden durch feuchte Hitze sterilisiert, um die Sterilität zu erhalten.
• Die Flüssigkeit wird nach der Trennung und Chromatographie einer Nanofiltration und Ultrafiltration unterzogen, die mit einer Sterilfiltration über einen 0,2μm-Filter abgeschlossen wird. Nach der Filtration wird die Flüssigkeit unter dem Schutz einer laminaren Strömung der Klasse A abgefüllt, um die mikrobielle Kontamination aus der Umgebung zu reduzieren. Die Gesamtflüssigkeit wird unter Einhaltung der Qualitätsstandards verschiedenen Tests unterzogen, einschließlich mikrobieller Grenzwerte und Endotoxin-Bewertungen. Wenn die Flüssigkeit diese Tests bestanden hat und von der Qualitätsabteilung genehmigt wurde, wird sie zur Abfüllung der Endprodukte weitergeleitet.
Formuliertes Produkt:
• Die Abfüllung des Endprodukts erfolgt in einem aseptischen Produktionsprozess ohne Sterilisation, wobei eine strenge B+A-Produktionsumgebung gewährleistet wird. Nach der Sterilisationsfiltration wird das Halbfertigprodukt gewonnen. Montage, Befüllung, Verschließen, automatisches Be- und Entladen und Lyophilisierung erfolgen mit vollautomatischen Anlagen und aseptischen Einweg-Abfüllsystemen in einer aseptischen Umgebung der Klasse A (PMS-System zur kontinuierlichen Umweltüberwachung). Nach der Gefriertrocknung wird die Versiegelung in einer C+A-Umgebung abgeschlossen. Die verpackten Produkte werden einer manuellen Sichtkontrolle, Etikettierung und weiteren Arbeitsschritten unterzogen. Sie werden erst nach der Inspektion ins Fertigwarenlager gebracht und freigegeben.
• Zu einer strengen aseptischen Prozessvalidierung gehört die regelmäßige Bestätigung der aseptischen Prozesskontrolle, einschließlich der Verwendung von aseptischen Nährmedien und/oder Produktersatzstoffen in APS (Mediensimulationsflaschen). APS bewertet alle aseptischen Vorgänge, die von der Sterilisation und Reinigung der Materialien bis hin zum Verschließen der Behälter durchgeführt werden. Die Trägersubstanzen werden einem Wachstumsförderungstest unterzogen, um zuverlässige Kulturergebnisse zu gewährleisten. Das Mikroben-Eindringungs-Experient dient als Verfahren zur Gewährleistung der Dichtigkeit des Verpackungssystems, um die Sterilität des Produkts während der Lagerung sicherzustellen.
• Qualitätskontrolle des fertigen Produkts: Der Sterilisationstest ist der letzte Schritt in einer Reihe von wesentlichen Kontrollmaßnahmen zur Gewährleistung der Sterilität. Die Sterilitätstests werden in einem aseptischen Isolator durchgeführt. Die Probenahme für die Sterilitätstests umfasst Produkte zu Beginn und am Ende der Chargenabfüllung als Referenz. Die Methoden der Sterilitätsanalyse halten sich streng an die Anforderungen des Arzneibuchs, wobei ATCC- und CMCC-zugelassene Stämme verwendet werden und die Anforderungen des Arzneibuchs für die Kultivierung biologischer Produkte eingehalten werden, um die Zuverlässigkeit der Ergebnisse zu gewährleisten.
Zusammenfassend lässt sich sagen, dass die GMP-gerechten Produkte von ACROBiosystems mit umfassenden aseptischen Schutzstrategien, strengen Qualitätskontrollen und unter Einhaltung der GMP-Vorschriften hergestellt werden. Diese aseptischen Verfahren unterstützen die Registrierung und Vermarktung von CGT-Produkten.
• Kontrolle externer Verunreinigungen bei der Herstellung von kritischen Materialien für Zell- und Gentherapien.
• Aseptische Schutzstrategien für die Produktion kritischer Materialien in der CGT
• Qualitätskontrollsystem für kritische Materialien in CGT
• Globales System zur Sicherung der Lieferkette für kritische Materialien in CGT
• Wie man die regulatorischen Anforderungen der Vereinigten Staaten für kritische Materialien in CGT besser erfüllen kann
• ...



This web search service is supported by Google Inc.
















 A-Z
A-Z





















